

Lab of Insect Genomics and Bioinformatics
Prof. Li Fei's group at Zhejiang University, HangZhou, China
Prof. Li Fei's group at Zhejiang University, HangZhou, China

By leveraging multi-omics data in entomology and advanced big data analytics, we've established a robust genetic analysis platform, InsectBase, which is the most extensive, comprehensive, and standardized data platform in the field of entomology , providing valuable resources for research in various fields of entomology.Concurrently, we have developed and amalgamated various genomic databases, including miR+Pathway, WaspBaseChiloDB, and iPathDB. Additionally, we've devised pertinent algorithms and computational workflows to fortify this platform. This infrastructure offers formidable technological backing for entomological research in the post-genomic epoch. Our findings have been disseminated in esteemed journals such as Nucleic Acids Research, Briefings in, Bioinformatics, Database, and Bioinformatics.
Employing bioinformatics and molecular biology techniques, we have undertaken genomic analyses of several pests affecting rice at the chromosome level, including the borer and rice planthopper. We've also investigated the genomes of invasive pests such as Cydia pomonella, Phenacoccus solenopsis, and Harmonia axyridis. Our investigations have unveiled the genetic traits inherent in these pests that facilitate their infestation, as well as the genomic basis of their adaptive evolution. Our findings have been featured in esteemed publications including Nature Communications, Molecular Ecology Resources, BMC Biology, and BMC Genomies. Additionally, we have received invitations to contribute reviews to Molecular Biology and other reputable journals.
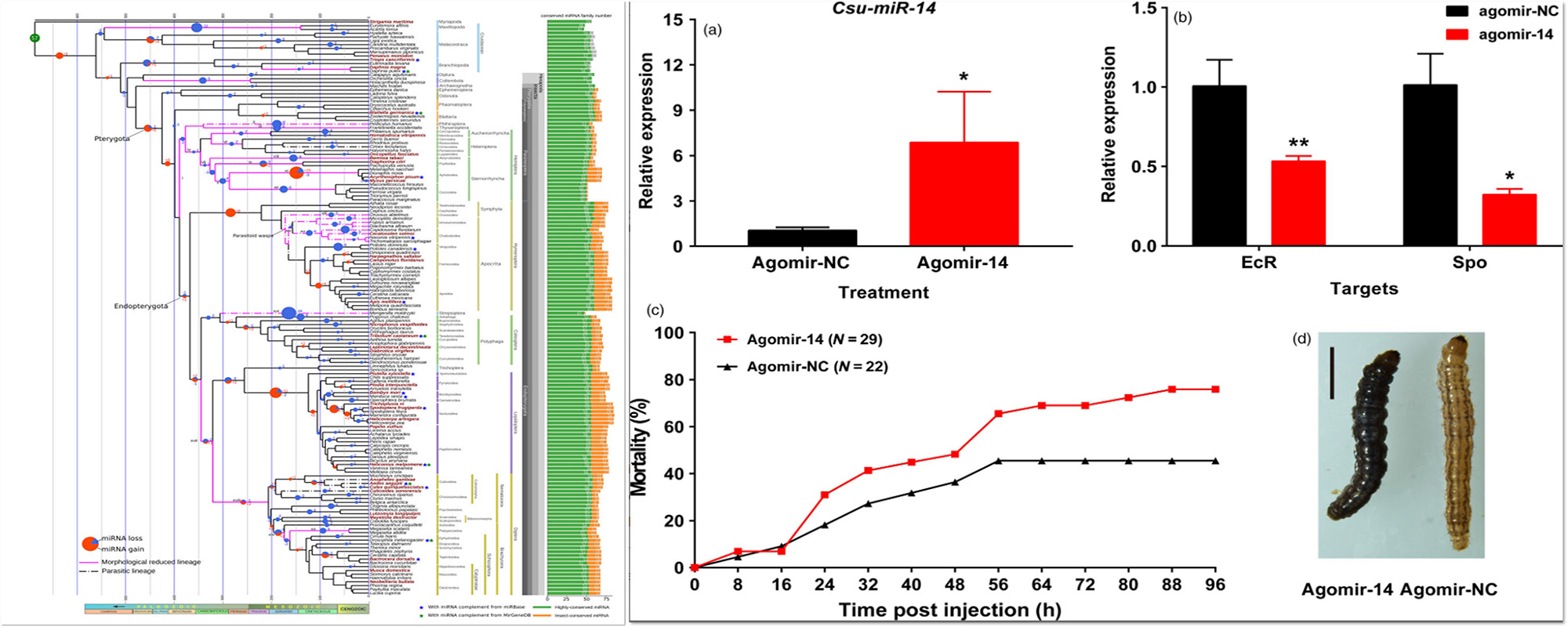
Non-coding RNAs play important regulatory functions at epigenetic and transcriptional levels. Our group has long been focusing on the rice screwworm and rice fly group as the research object, and is committed to digging out the key regulatory genes from the histological big data, and exploring the application prospect of microRNAs and other non-coding RNAs in the metamorphosis development, super-typical differentiation, and reproduction regulation through the study of the function of microRNAs and other RMAs in the green pest control, and the related research has been published in Nature communications, Plant Biotechnology Journal, PloS Genetics, RNA Biology, Genome Biology Evolution.

Micro CT, as a mature imaging technique, has found widespread applications across various scientific disciplines. Its primary advantage lies in the ability to monitor internal structures of objects with resolutions as fine as several hundred nanometers while preserving the original morphology of the specimens. Our Lab has developed a process for reconstructing internal models of insects using Micro CT. This process has been successfully employed to construct three-dimensional models of the central nervous system, muscle tissues, digestive tract, and reproductive system within the brown planthopper. Furthermore, this method can be integrated with genetic mutation studies, allowing for precise quantitative analysis of morphological changes and providing richer phenotypic information for insect development.

Published on: 2025年1月29日
Thanks to the fast development of sequencing techniques and bioinformatics tools, sequencing the genome of an insect species for specific research purposes has become an increasingly popular practice. Insect genomes not only provide sets of gene sequences but also represent a change in focus from reductionism to systemic biology in the field of entomology. Using insect genomes, researchers are able to identify and study the functions of all members of a gene family, pathway, or gene network associated with a trait of interest. Comparative genomics studies provide new insights into insect evolution, addressing long-lasting controversies in taxonomy. It is also now feasible to uncover the genetic basis of important traits by identifying variants using genome resequencing data of individual insects, followed by genome-wide association analysis. Here, we review the current progress in insect genome sequencing projects and the application of insect genomes in uncovering the phylogenetic relationships between insects and unraveling the mechanisms of important life-history traits. We also summarize the challenges in genome data sharing and possible solutions. Finally, we provide guidance for fully and deeply mining insect genome data.
Read more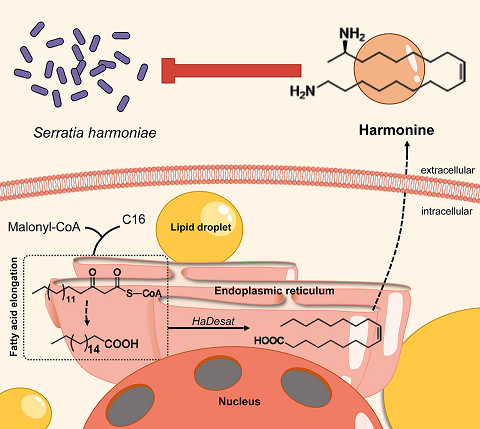
Published on: 2024年12月30日
Once prized for its use in biological pest control, the harlequin ladybird (Harmonia axyridis ) has become an invasive pest in nonnative regions, outcompeting local ladybird species. Here, we found that the harlequin ladybird safely harbors Serratia harmoniae , a highly pathogenic bacterium that causes severe mortality in other ladybird species. The harlequin ladybird’s tolerance to the pathogen is attributed to the defense alkaloid harmonine. Silencing three key genes in the harmonine biosynthesis pathway—Spidey , Sca2 , and Desat —reduced the production of harmonine, leading to increased bacterial levels and increased mortality. Penicillin treatment reversed this effect, reducing S. harmoniae content and increasing host survival. This symbiotic host–pathogen relationship confers an intraguild predation advantage to the harlequin ladybird.
Read more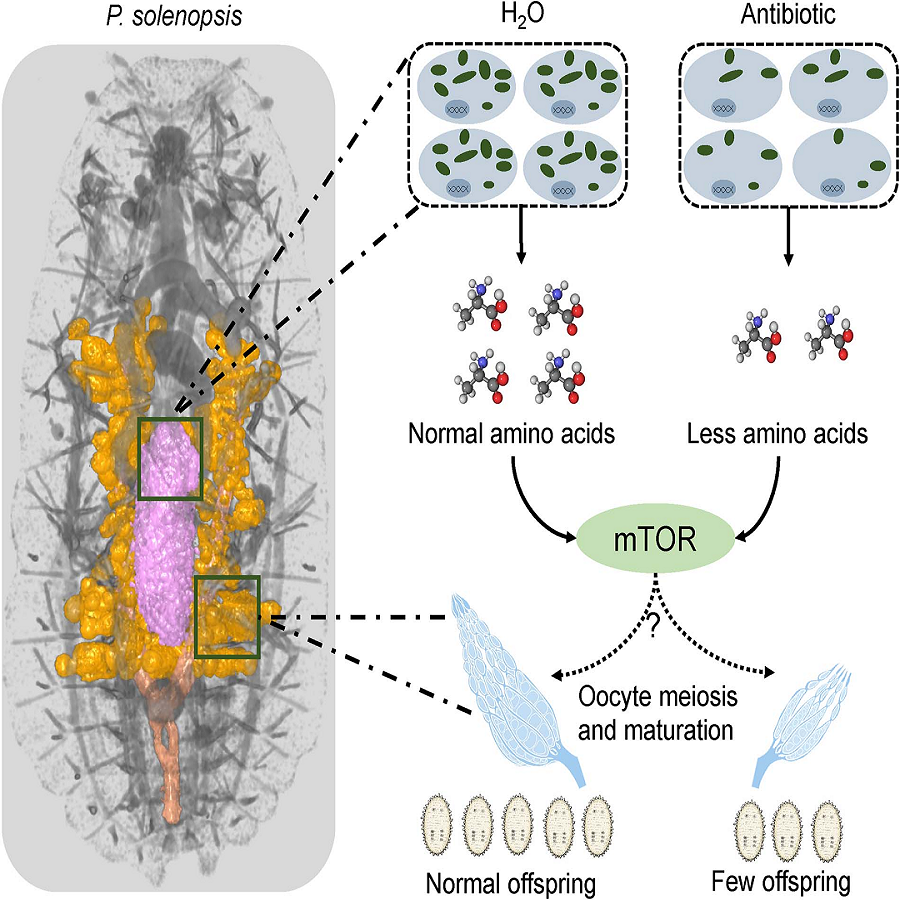
Published on: 2024年3月22日
The endosymbiont, “Candidatus Tremblaya phenacola” (T. phenacola PSOL), persisted throughout the complete life cycle of female hosts and was more active during oviposition, whereas there was a significant decline in abundance after pupation in males. A comprehensive analysis of amino acid metabolic pathways demonstrated complementarity between the host and endosymbiont metabolism. Elimination of T. phenacola PSOL through antibiotic treatment significantly decreased P. solenopsis fecundity. Weighted gene coexpression network analysis demonstrated a correlation between genes associated with essential amino acid synthesis and those associated with host meiosis and oocyte maturation. Moreover, altering endosymbiont abundance activated the host mechanistic target of rapamycin pathway, suggesting that changes in the amino acid abundance affected the host reproductive capabilities via this signal pathway. Taken together, these findings demonstrate a mechanism by which the endosymbiont T. phenacola PSOL contributed to high fecundity in P. solenopsis and provide new insights into nutritional compensation and coevolution of the endosymbiotic system.
Read more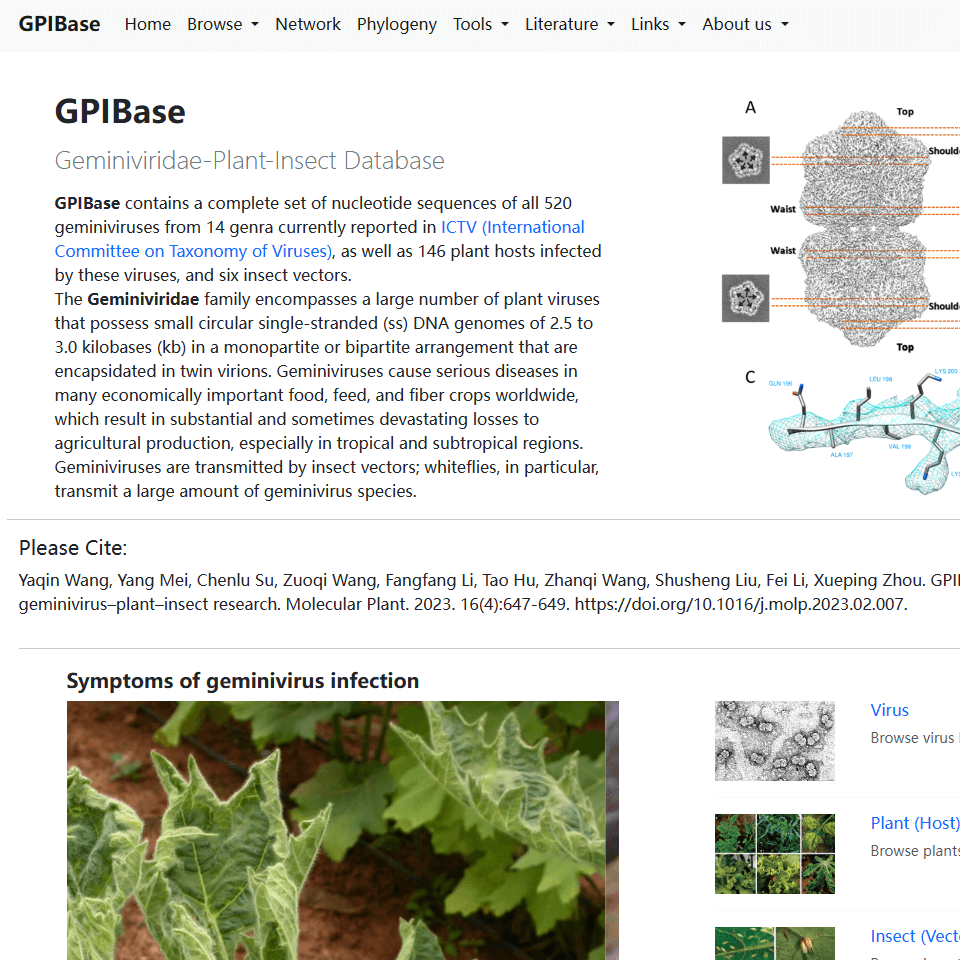
Published on: 2023年4月3日
Here, we constructed a database by integrating the genomes and gene information of geminiviruses, their plant hosts, and insect vectors, named GPIBase (http://gpi.geminiviridae.com/). This database contains a complete set of nucleotide sequences from representative members of all 520 geminivirus species (one per species) and 119 betasatellites currently accepted by the International Committee on Taxonomy of Viruses, as well as 146 plant hosts infected by these viruses and six insect vectors. Interactions between plants, insects, and viruses are analyzed and presented, and functions of searching, genome browser, and phylogenetic analysis are also included. In general, GPIBase provides a widely accessible, simple, user-friendly, and knowledge-oriented platform. Users can retrieve information related to geminiviruses for studying tripartite connections between geminiviruses, plants, and insects
Read more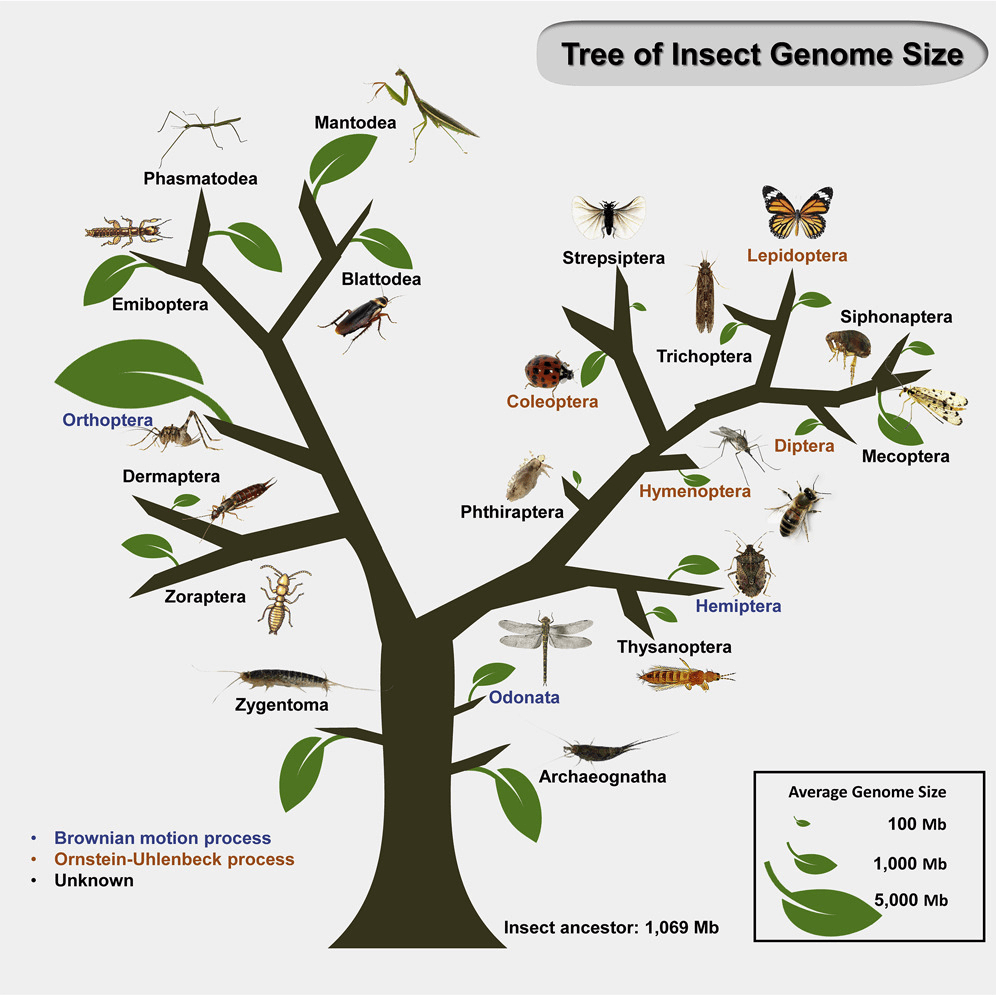
Published on: 2022年8月3日
Genome size (GS) can vary considerably between phylogenetically close species, but the landscape of GS changes in insects remain largely unclear. To better understand the specific evolutionary factors that determine GS in insects, we examined flow cytometry-based published GS data from 1,326 insect species, spanning 700 genera, 155 families, and 21 orders. Model fitting showed that GS generally followed an Ornstein–Uhlenbeck adaptive evolutionary model in Insecta overall. Ancestral reconstruction indicated a likely GS of 1,069 Mb, suggesting that most insect clades appeared to undergo massive genome expansions or contractions. Quantification of genomic components in 56 species from nine families in four insect orders revealed that the proliferation of transposable elements contributed to high variation in GS between close species, such as within Coleoptera. This study sheds lights on the pattern of GS variation in insects and provides a better understanding of insect GS evolution.
Read more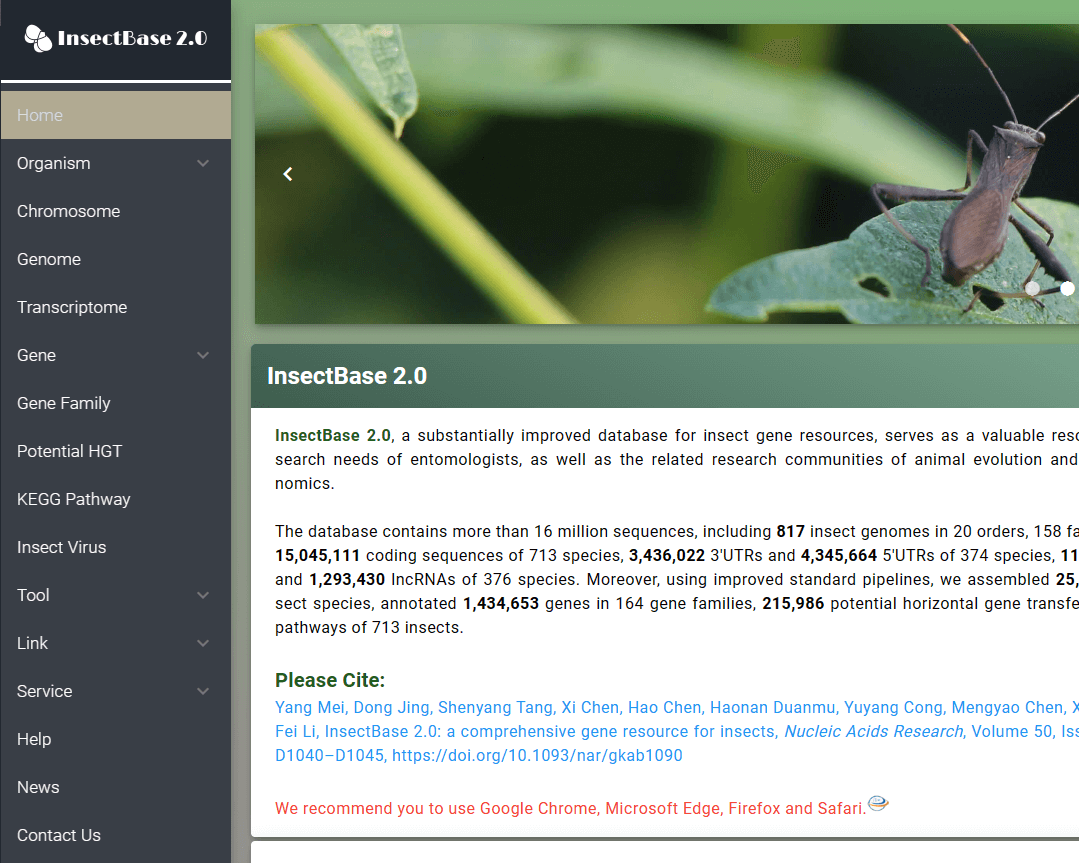
Published on: 2022年1月7日
We present an updated database, InsectBase 2.0 (http://v2.insect-genome.com/), covering 815 insect genomes, 25 805 transcriptomes and >16 million genes, including 15 045 111 coding sequences, 3 436 022 3'UTRs, 4 345 664 5'UTRs, 112 162 miRNAs and 1 293 430 lncRNAs. In addition, we used an in-house standard pipeline to annotate 1 434 653 genes belonging to 164 gene families; 215 986 potential horizontally transferred genes; and 419 KEGG pathways. InsectBase 2.0 serves as a valuable platform for entomologists and researchers in the related communities of animal evolution and invertebrate comparative genomics.
Read more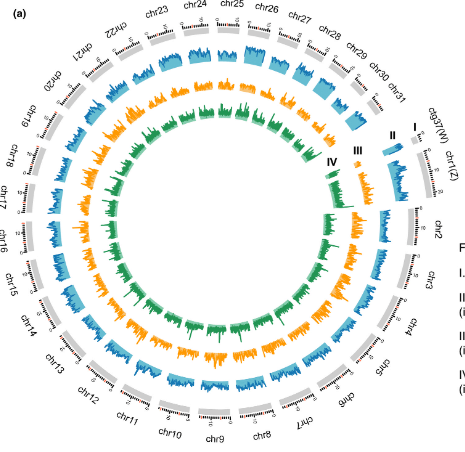
Published on: 2020年5月2日
The fall armyworm, scientifically known as Spodoptera frugiperda, is a destructive lepidopteran insect pest that inflicts significant economic losses. Over recent years, it has rapidly spread worldwide, but the mechanisms behind its swift dispersal have remained unclear. To address this, we present a high-quality genome assembly of the fall armyworm, referred to as the ZJ-version. This genome was generated using advanced technologies such as PacBio and Hi-C, with the sample collected from Zhejiang province, China, exhibiting high heterozygosity. The ZJ-version has a genome size of 486 Mb, comprising 361 contigs with an N50 of 1.13 Mb. Further Hi-C scaffolding led to the assembly of 31 chromosomes and a portion of the W chromosome, achieving a chromosome-level genome with a scaffold N50 of 16.3 Mb. Identification of sex chromosomes was achieved through genome resequencing of a single male and female pupa.
Read more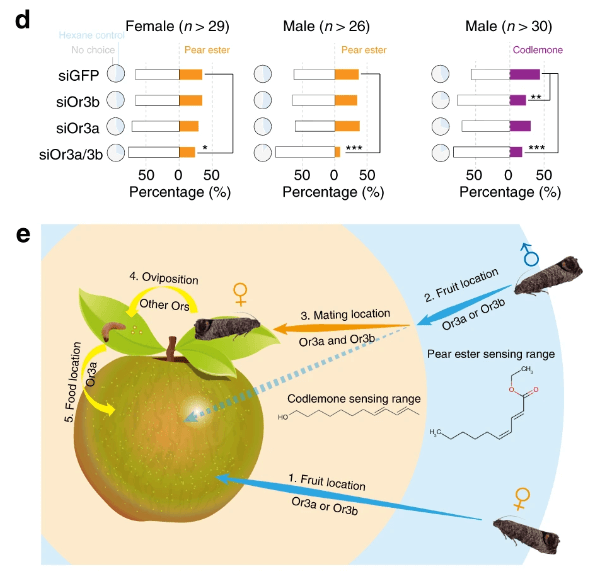
Published on: 2019年9月17日
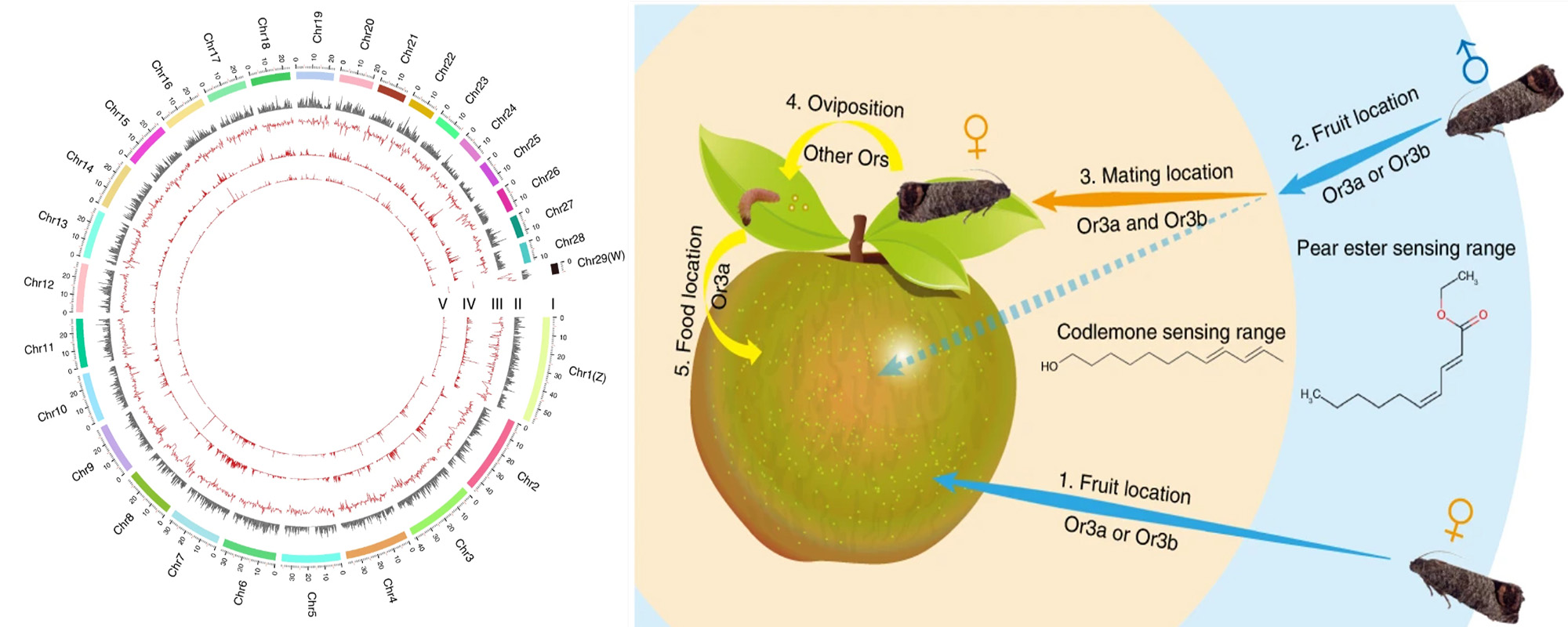
The codling moth Cydia pomonella, a major invasive pest of pome fruit, has spread around the globe in the last half century. We generated a chromosome-level scaffold assembly including the Z chromosome and a portion of the W chromosome. This assembly reveals the duplication of an olfactory receptor gene (OR3), which we demonstrate enhances the ability of C. pomonella to exploit kairomones and pheromones in locating both host plants and mates. Genome-wide association studies contrasting insecticide-resistant and susceptible strains identify hundreds of single nucleotide polymorphisms (SNPs) potentially associated with insecticide resistance, including three SNPs found in the promoter of CYP6B2. RNAi knockdown of CYP6B2 increases C. pomonella sensitivity to two insecticides, deltamethrin and azinphos methyl. The high-quality genome assembly of C. pomonella informs the genetic basis of its invasiveness, suggesting the codling moth has distinctive capabilities and adaptive potential that may explain its worldwide expansion.
Read more